

Sequence analysis of pol gene of HIV-1 strains in Kunming, Yunnan Province
-
摘要:
目的 了解云南省昆明市人类免疫缺陷病毒1型(human immunodeficiency virus type 1,HIV-1)的亚型分布特征及其pol结构基因的变异特征。 方法 随机抽取2015—2019年昆明市抗病毒治疗半年以上的样本390例,收集其流行病学信息,血浆提纯HIV-1 RNA,采用巢式聚合酶链式反应扩增HIV-1 pol基因,应用Genotyping Tool、BLAST在线分型工具及MEGA 6.06软件进行基因分型,运用MEGA Distance程序计算基因距离,Entropy程序分析氨基酸多态性,进而对HIV-1流行株的pol基因序列特征进行分析。 结果 成功扩增获得pol基因序列318例,云南省昆明市的HIV-1亚型主要为B亚型占比3.1%,C亚型占比4.4%,CRF01_AE占比26.1%,CRF07_BC占比20.4%,CRF08_BC占比42.8%,其他亚型占比3.1%。各个亚型在性别、年龄及感染途径的构成上差异均有统计学意义(均有P < 0.001)。CRF01_AE、CRF07_BC、CRF08_BC的基因距离分别为(0.018±0.008)、(0.015±0.006)和(0.006±0.004)bp,差异均有统计学意义(均有P < 0.01)。CRF01_AE亚型中静脉药瘾感染者与性接触感染者中共有45个位点的氨基酸差异存在统计学意义(均有P < 0.05),异性性传播与同性性传播中共156个位点有差异。 结论 昆明市HIV-1亚型分布复杂多样,以CRF08_BC为主,传播途径以异性性传播为主,CRF01_AE亚型为昆明地区变异程度较高的流行株,不同感染途径毒株的氨基酸突变位点存在差异,应密切监视其变化。 -
关键词:
- 人类免疫缺陷病毒1型 /
- 亚型分布 /
- 基因距离 /
- 氨基酸变异
Abstract:Objective To investigate the distribution of human immunodeficiency virus type 1(HIV-1) subtypes and the variation of pol gene in Kunming, Yunnan Province. Methods Randomly selected 390 samples with HAART for more than half a year in Kunming from 2015 to 2019, and collected the epidemiological information. Purified the HIV-1 RNA from plasma and amplified the HIV-1 pol gene by nested polymerase chain reaction. Online analysis tools like Genotyping, BLAST and MEGA 6.06 software were used to determine strain subtypes. Distance program of MEGA was used to calculate the gene distance, and Entropy program was used to analyze the amino acid polymorphism, so as to analyze the pol gene sequence characteristics of HIV-1 strains. Results A total of 318 pol gene sequences were successfully amplified. The major subtypes of HIV-1 strains in Kunming were B subtype(3.1%), C subtype(4.4%), CRF01_AE(26.1%), CRF07_BC(20.4%), CRF08_ BC(42.8%) and other subtypes (3.1%). There were statistically significant differences in the distribution of HIV-1 genotypes in sex, age and infection routes (all P < 0.001).The genetic distances of pol gene for CRF01_AE, CRF07_BC and CRF08_BC were (0.018±0.008), (0.015±0.006) and (0.006±0.004) bp (all P < 0.01). In CRF01_ AE, there were 45 different sites in amino acid sequences between intravenous drug addicts and sexual contacts and 156 different sites between hetrosexual contacts and homosexual contacts, and the differences were sitatistically significant (P < 0.05). Conclusion There are complex and diverse HIV-1 subtypes in Kunming, and CRF08_BC is the predominant epidemic subtype. The main route of infection is heterosexual transmission. CRF01_ AE subtype is an epidemic strain with high degree of variation in Kunming. There are differences in amino acid mutation sites of strains from different infection routes, which should be closely monitored. -
人类免疫缺陷病毒1型(human immunodeficiency virus type 1, HIV-1) 属于单股正链RNA反转录病毒科慢病毒属。HIV的基因具有高度变异性,其高度变异性使得病毒能够避免被宿主清除或逃离抗病毒药物的作用,其中HIV-1广泛遗传变异的原因包括:病毒逆转录酶缺乏校对活性[1],病毒粒子在复制过程中快速周转[2-3],以及感染细胞中发生基因重组等[4]。由于现有的全长序列相对较少,且受到测序成本等多种因素的限制,故而选择合适的遗传区域进行分析时需仔细考虑。目前大多数的研究[5]区域集中于编码胞膜蛋白的env基因,少部分为编码核心蛋白的gag区,而HIV-1的pol基因编码蛋白水解酶、逆转录酶和整合酶,对HIV-1的复制、前病毒的整合、增殖和药物敏感性都具有重要意义[6],pol基因作为抗病毒治疗药物的主要作用靶点,其基因的变异对治疗成败至关重要。pol基因中,HIV-1亚型之间核苷酸差异约为9%~11%[7],属于HIV基因组中较为保守的片段。目前在基因型耐药性检测的临床背景下,pol基因的蛋白酶区和逆转录酶区正在产生大量的序列数据,这些片段组成了庞大的序列数据库,为HIV分子流行病学的研究提供基础,且利用pol进行基因分型能够较好地识别一些重组毒株[8]。据报道[9-10],只要所使用的片段足够长和可变,就能成功基于pol区蛋白酶和逆转录酶片段对HIV-1进行基因分型。
中国已报道了多种HIV-1亚型包括A、B、B’、C、D、F和多种流行重组型(circulating recobinant forms, CRFs)包括CRF01_AE、CRF07_BC、CRF08_BC等[11]。而云南省由于其特殊的地理位置一直是AIDS的高流行地区。因此,应用分子流行病学的方法了解分析云南省HIV-1 pol区基因序列特征,可初步掌握云南省昆明市的HIV-1亚型分布情况,为今后的进一步分析提供参考。
1. 对象与方法
1.1 研究对象
随机抽取经Western Blot实验确证为HIV-1阳性,并于2015—2019年在云南省传染病医院接受抗病毒治疗半年以上且血浆检测HIV-1病毒载量>1 000 copies/ml的HIV/AIDS患者共390例,本研究样本采集遵循知情同意原则。
1.2 实验方法
1.2.1 核酸提取及PCR扩增
将样本从-80 ℃的冰箱中取出并在室温下自然解冻,采用雅培Abbott m2000sp自动化样本核酸提纯系统和配套试剂提取HIV-1 RNA,具体操作按试剂盒说明书进行。采用巢式聚合酶链式反应扩增HIV-1 pol区编码蛋白酶及反转录酶的片段,全长约1 200 bp。具体的引物及反应体系参照文献[10]。
1.2.2 扩增产物鉴定及测序
第二轮PCR扩增产物采用1%的琼脂糖凝胶电泳鉴定,将扩增阳性的产物送至北京诺赛基因测序公司进行测序。
1.2.3 序列分析
对所得的测序片段使用Conting Express 6.0软件进行剪切和拼接后,采用BioEdit软件将所得的样本序列与标准序列进行比对校准。序列的初步分型采用美国国家生物技术信息中心(national center of biotechnology information, NCBI)的在线分型工具Genotyping Tool进行,同时使用Los Alamos HIV数据库中的HIV BLAST工具进行分型鉴定,将本研究得到的序列与Los Alamos HIV数据库中的标准序列进行比对,用MEGA 6.06软件构建Neighbor-joining系统进化树(重复运算1 000次)进行分型比对,将bootstrap值>70%定义为流行簇。用Distance程序计算基因离散率。用Entropy软件对序列的氨基酸多态性进行分析。
1.3 统计学方法
使用Excel 2010软件对样本的人口学资料进行统计,使用SPSS 24.0软件对数据进行统计分析,分类资料及构成比采用χ2检验或Fisher确切概率法检验,若符合正态分布, 多组样本均数的比较则采用方差分析,两两之间比较采用SNK检验,若不符合,则采用秩和检验。检验水准α=0.05。
2. 结果
2.1 HIV-1亚型人口学分布特征
共收集到390例符合条件的血浆样本,成功扩增获得pol基因序列318例,扩增阳性率为81.5%(318/390)。结果显示,不同亚型在性别、年龄及感染途径的构成上有差别(均有P < 0.01)。其中CRF08_BC为最主要的亚型,占42.8%,本研究中的HIV/AIDS患者主要为中青年男性,异性性传播为最主要的传播途径。见表 1。
表 1 2015—2019年昆明市318例HIV/AIDS患者人口学特征及基因亚型分布情况[n(%)]Table 1. Demographic characteristics and gene subtype distribution of 318 HIV/AIDS patients in Kunming City from 2015 to 2019 [n(%)]变量 例数(n=318) B C CRF01_AE CRF07_BC CRF08_BC 其他 χ2值 P值 性别 29.381 < 0.01 男 224(70.4) 9(4.0) 8(3.6) 75(33.5) 42(18.8) 82(36.6) 8(3.6) 女 94(29.6) 1(1.0) 6(6.4) 8(8.5) 23(24.4) 54(57.4) 2(2.1) 年龄(岁) 42.344 < 0.01 < 20 20(6.3) - 2(10.0) 1(5.0) 5(25.0) 12(60.0) - 20~ < 40 153(48.1) 6(3.9) 8(5.2) 57(37.3) 32(20.9) 42(27.5) 8(5.2) 40~ < 60 135(42.5) 4(3.0) 3(2.2) 25(18.5) 26(19.3) 75(55.6) 2(1.5) ≥60 10(3.1) - 1(10.0) - 2(20.0) 7(70.0) - 感染途径 113.846 < 0.01 静脉吸毒 86(27.0) - 3(3.5) 7(8.1) 19(22.1) 57(66.3) - 异性性传播 120(37.3) 4(3.3) 7(5.8) 25(20.8) 25(20.8) 56(46.7) 3(2.5) 同性性传播 62(19.5) 4(6.5) - 38(61.3) 12(19.4) 2(3.2) 6(9.7) 母婴传播 19(6.0) - 2(10.5) 1(5.3) 5(26.3) 11(57.9) - 其他 31(9.7) 2(6.5) 2(6.5) 12(38.7) 4(12.9) 10(32.3) 1(3.2) 合计 318(100.0) 10(3.1) 14(4.4) 83(26.1) 65(20.4) 136(42.8) 10(3.1) 注:其他亚型(例数)依次为A(2)、CRF55_01B(6)、CRF78_cpx(1)以及CRF86_BC(1)。 2.2 主要亚型的系统进化树分析
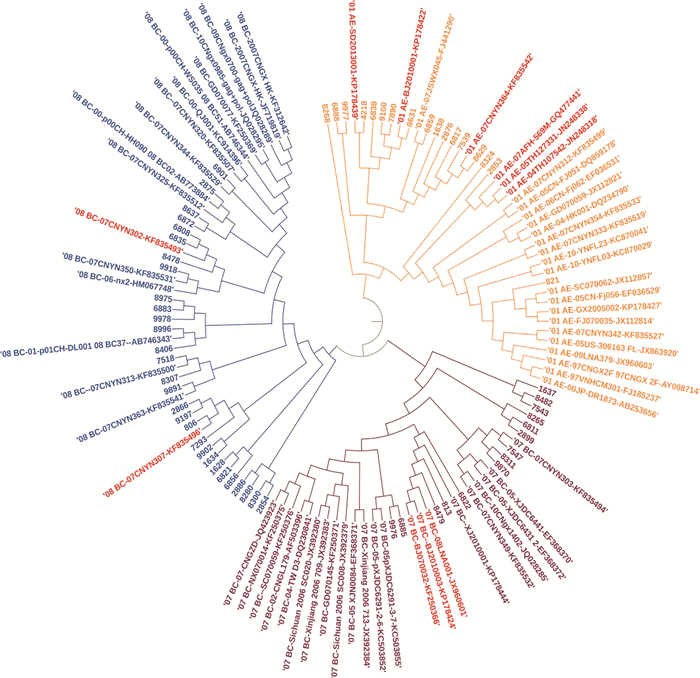
按照分层随机抽样的方法在扩增成功的样本中分别抽取17例CRF01_AE、14例CRF07_BC以及29例CRF08_BC共计60例样本构建系统进化树。从图 1中可以看出pol基因片段在进化树中分为两个大的分支,分别为CRF01_AE分支和CRF07_BC/CRF08_BC分支,在CRF01_AE亚型中,其中两株传播途径为同性性传播的男性样本与来自中国山东省的标准株01_AE-SD2013001-KP178439聚在一起,一株传播途径为静脉药瘾的男性样本与来自泰国的两株标准株01_AE-05TH127331-JN248338和01_AE-04TH107542-JN248318聚在一起;在CRF07_BC亚型中,一株同性性传播的样本与来自北京的标准株07_BC-BJ070032-KF250366和07_BC-08LNA001-JX960601聚在一起;在CRF08_BC亚型中,两株均为静脉药瘾感染的男性样本与来自云南省的标准株08_BC-07CNYN302-KF835493聚在一起。
 图 1 昆明市HIV-1 pol基因序列系统进化树分析Figure 1. Phylogenetic tree analysis of HIV-1 pol gene sequence in Kunming
图 1 昆明市HIV-1 pol基因序列系统进化树分析Figure 1. Phylogenetic tree analysis of HIV-1 pol gene sequence in Kunming2.3 pol区基因距离分析
基因距离分析结果表明CRF01_AE的基因距离最大而CRF08_BC的基因距离最小。经方差分析显示各亚型间基因距离的差距有统计学意义(均有P < 0.01),经事后使用SNK法两两比较发现,三种重组型+++间的基因距离差异均有统计学意义(均有P < 0.01)。见表 2。
表 2 2015—2019年昆明市318例HIV/AIDS患者HIV-1 pol基因的三种主要亚型基因距离Table 2. Gene distances of three main subtypes of HIV-1 pol gene in 318 HIV/AIDS patients in Kunming from 2015 to 2019HIV-1亚型 例数(n=276) 基因距离(x±s, bp) F值 P值 CRF01_AE 75 0.018±0.008 10 739.856 < 0.001 CRF07_BC 58 0.015±0.006 CRF08_BC 143 0.006±0.004 2.4 CRF01_AE亚型的氨基酸多态性分析
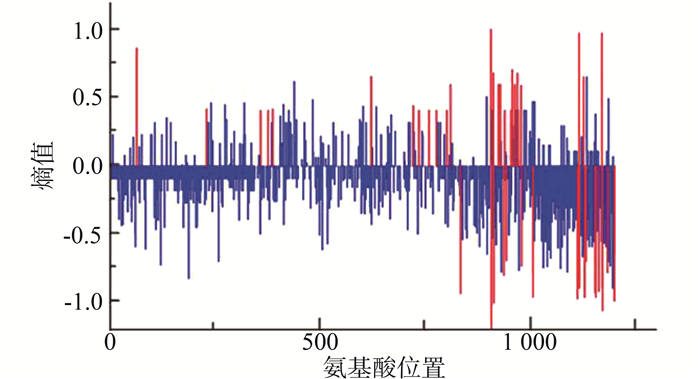
将CRF01-AE按照传播途径分为静脉药瘾和性传播途径(包括同性及异性性传播)两组,以静脉药瘾感染的一组为背景序列,性传播途径感染的为分析序列,红色竖线表示两组序列氨基酸分布差异有统计学意义(P < 0.05)的位点,结果显示两组样本共有45个位点的多态性存在差异(均有P < 0.05),其中感染途径为静脉吸毒的序列相对于性传播途径氨基酸多态性增加的位点有26个,总体上看背景序列的多态性稍大于分析序列,在这些位点上氨基酸分布类型背景序列更为丰富。见图 2。
 图 2 CRF01_AE亚型两组不同感染途径序列氨基酸多态性分析Figure 2. Amino acid poly morphism analysis of different infection routes in two groups of CRF01_AE subtype
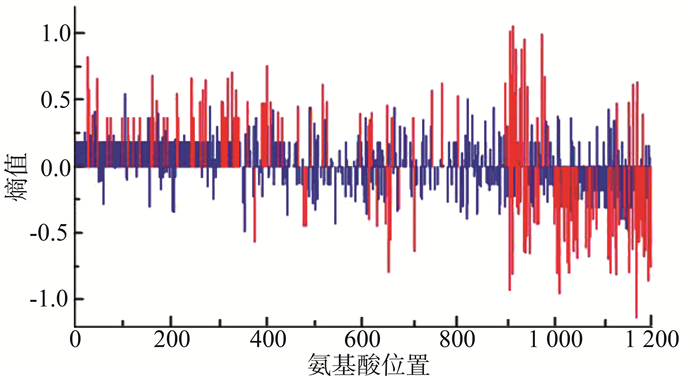
图 2 CRF01_AE亚型两组不同感染途径序列氨基酸多态性分析Figure 2. Amino acid poly morphism analysis of different infection routes in two groups of CRF01_AE subtype进一步分析性传播途径中CRF01_AE的氨基酸多态性结果显示,同性性传播及异性性传播两组中共有156个位点的氨基酸多态性存在差异(均有P < 0.05),其中熵值>0的共有83个,即感染途径为异性性传播的患者氨基酸多态性大于同性性传播的患者。见图 3。
 图 3 CRF01_AE亚型中异性性传播与同性性传播患者序列氨基酸多态性分析Figure 3. Sequence amino acid polymorphism analysis of heterosexual and homosexual transmission patients in CRF01_AE subtype
图 3 CRF01_AE亚型中异性性传播与同性性传播患者序列氨基酸多态性分析Figure 3. Sequence amino acid polymorphism analysis of heterosexual and homosexual transmission patients in CRF01_AE subtype3. 讨论
过去30年里,几乎所有地区和风险群体的HIV-1基因型在全国范围内的分布都发生了巨大的变化,与目前中国大部分地区流行的亚型为CRF01_AE不同[12],此次研究中分布占比较高的三种亚型依次为CRF08_BC、CRF01_AE和CRF07_BC,与之前云南省亚型分布的研究[13-14]结论一致。云南省因其特殊的地理位置,对全国其他地区的HIV流行有着直接或间接的影响[15]。由于20世纪八九十年代,B’亚型和C亚型在云南省德宏傣族景颇族自治州静脉吸毒人群中的流行[16],为各种B’/C重组形式的产生提供了机会,其中就包括目前在云南省广泛流行的CRF08_BC和CRF07_BC[17]。此外,本研究中还出现了6例CRF55_01B,其中5例为同性性传播,1例为异性性传播,该亚型于2013年在中国湖南省长沙市和广东省东莞市的MSM中首次报道,它由CRF01_AE和B6亚型组成,主要分布在广东省及周边省份[18],在云南省的发现提示CRF55_01B已有从东南沿海城市向西南蔓延的趋势。
云南省目前最主要的传播途径为性传播,其次为静脉药瘾,与以往其他省份研究[19]结果一致。在CRF08_BC中仍以静脉药瘾传播为主,这可能是云南省西南部边境比邻东南亚国家,而CRF08_BC起源于此,是中国静脉吸毒传播的主要亚型毒株[20],随着时间推移,性传播途径在CRF08_BC中的占比逐渐增加,提示HIV-1的传播已经逐渐由高危人群向普通人群扩散。除CRF08_BC外,其他几种亚型均以性传播途径为主,在CRF01_AE中同性性传播占比高达61.3%。结合系统进化树,同性性传播的几株样本靠近来自山东省、北京市的参考株。近年来,在许多经济发展迅速且人口流动较大的城市中MSM的比例逐渐扩大,Zhang等[21]2015年的研究结果显示,CRF01_AE最近已取代B亚型成为中国MSM主要流行的亚型,因此应针对该人群进行重点干预。而静脉药瘾的几株样本靠近来自泰国和中国云南省的参考株,这与该传播途径在云南省的起源是相符的。
对基因距离的分析显示,距离越大其变异程度越高,说明该基因型出现的时间越早,流行的时间越长。CRF01_AE作为重组毒株中的优势毒株,其变异程度较高,该亚型于1990年首次在中国云南省的暗娼人群中发现,最初被称作“E亚型”[22],由于传播途径由早期的静脉吸毒及血液传播逐渐演变为以性接触为主,导致该亚型在全国范围内广泛流行[23],并且昆明市流动人口较多且多为青壮年,处于性行为活跃期容易发生高危性行为,交叉感染成为了CRF01_AE发生变异的推动因素。
进一步对变异程度较高的CRF01_AE亚型进行氨基酸多态性分析发现,静脉吸毒的人群比性接触感染的氨基酸多态性增加,在性传播途径中,异性性传播又比同性性传播的氨基酸分布类型更为丰富。有研究[24]表明,吸毒人群在CRF01_AE亚型中的分布比例持续上升,由于静脉吸毒感染方式进入时间较早,随着流行时间的延长从而造成其氨基酸类型更为多样。而在性接触感染人群中,异性性传播相较同性性传播流行时间更长,且人口基数大,传染源较分散,优势毒株在人群中的流行交叉更为频繁,导致其氨基酸更易发生变异[25]。
综上所述,目前昆明市具有多种HIV-1亚型并存的复杂特征,以异性性传播为主,对极易产生基因变异的CRF01_AE亚型应密切监测,为制定有效地防控及干预措施提供依据。
-
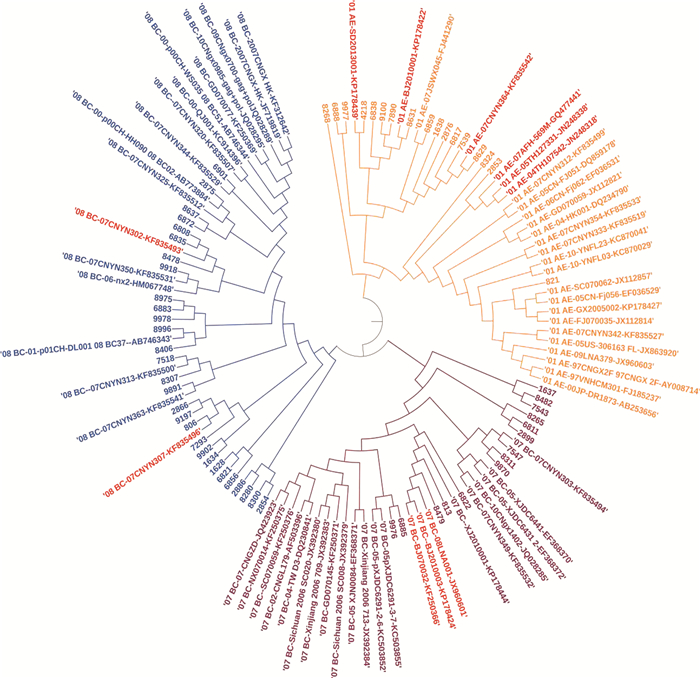
图 1 昆明市HIV-1 pol基因序列系统进化树分析
Figure 1. Phylogenetic tree analysis of HIV-1 pol gene sequence in Kunming
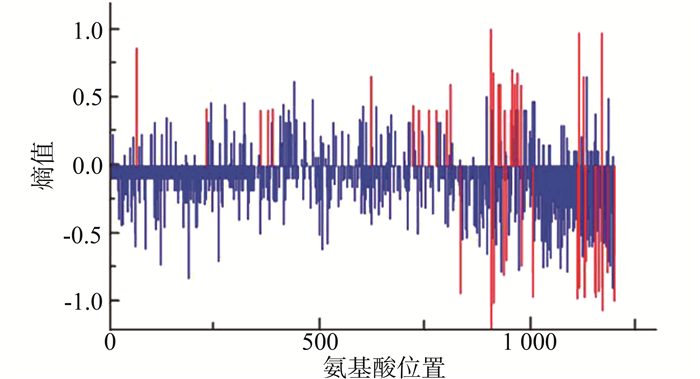
图 2 CRF01_AE亚型两组不同感染途径序列氨基酸多态性分析
Figure 2. Amino acid poly morphism analysis of different infection routes in two groups of CRF01_AE subtype
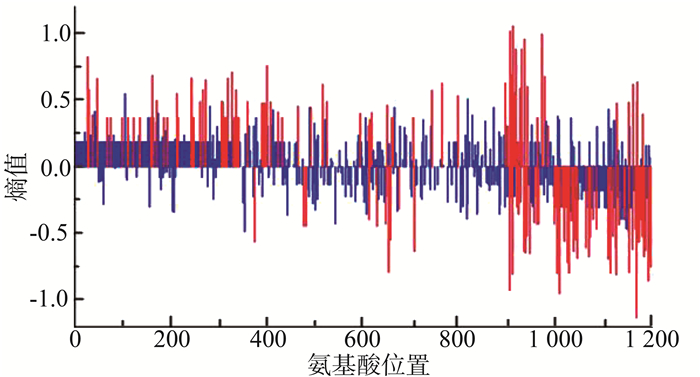
图 3 CRF01_AE亚型中异性性传播与同性性传播患者序列氨基酸多态性分析
Figure 3. Sequence amino acid polymorphism analysis of heterosexual and homosexual transmission patients in CRF01_AE subtype
表 1 2015—2019年昆明市318例HIV/AIDS患者人口学特征及基因亚型分布情况[n(%)]
Table 1. Demographic characteristics and gene subtype distribution of 318 HIV/AIDS patients in Kunming City from 2015 to 2019 [n(%)]
变量 例数(n=318) B C CRF01_AE CRF07_BC CRF08_BC 其他 χ2值 P值 性别 29.381 < 0.01 男 224(70.4) 9(4.0) 8(3.6) 75(33.5) 42(18.8) 82(36.6) 8(3.6) 女 94(29.6) 1(1.0) 6(6.4) 8(8.5) 23(24.4) 54(57.4) 2(2.1) 年龄(岁) 42.344 < 0.01 < 20 20(6.3) - 2(10.0) 1(5.0) 5(25.0) 12(60.0) - 20~ < 40 153(48.1) 6(3.9) 8(5.2) 57(37.3) 32(20.9) 42(27.5) 8(5.2) 40~ < 60 135(42.5) 4(3.0) 3(2.2) 25(18.5) 26(19.3) 75(55.6) 2(1.5) ≥60 10(3.1) - 1(10.0) - 2(20.0) 7(70.0) - 感染途径 113.846 < 0.01 静脉吸毒 86(27.0) - 3(3.5) 7(8.1) 19(22.1) 57(66.3) - 异性性传播 120(37.3) 4(3.3) 7(5.8) 25(20.8) 25(20.8) 56(46.7) 3(2.5) 同性性传播 62(19.5) 4(6.5) - 38(61.3) 12(19.4) 2(3.2) 6(9.7) 母婴传播 19(6.0) - 2(10.5) 1(5.3) 5(26.3) 11(57.9) - 其他 31(9.7) 2(6.5) 2(6.5) 12(38.7) 4(12.9) 10(32.3) 1(3.2) 合计 318(100.0) 10(3.1) 14(4.4) 83(26.1) 65(20.4) 136(42.8) 10(3.1) 注:其他亚型(例数)依次为A(2)、CRF55_01B(6)、CRF78_cpx(1)以及CRF86_BC(1)。  下载: 导出CSV
下载: 导出CSV
表 2 2015—2019年昆明市318例HIV/AIDS患者HIV-1 pol基因的三种主要亚型基因距离
Table 2. Gene distances of three main subtypes of HIV-1 pol gene in 318 HIV/AIDS patients in Kunming from 2015 to 2019
HIV-1亚型 例数(n=276) 基因距离(x±s, bp) F值 P值 CRF01_AE 75 0.018±0.008 10 739.856 < 0.001 CRF07_BC 58 0.015±0.006 CRF08_BC 143 0.006±0.004
下载: 导出CSV
-
[1] Preston BD, Dougherty JP. Mechanisms of retroviral mutation[J]. Trends Microbiol, 1996, 4(1): 16-21. DOI: 10.1016/0966-842x(96)81500-9. [2] Ho DD, Neumann AU, Perelson AS, et al. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection[J]. Nature, 1995, 373(6510): 123-126. DOI: 10.1038/373123a0. [3] Wei X, Ghosh SK, Taylor ME, et al. Viral dynamics in human immunodeficiency virus type 1 infection[J]. Nature, 1995, 373(6510): 117-122. DOI: 10.1038/373117a0. [4] Robertson DL, Hahn BH, Sharp PM. Recombination in AIDS viruses[J]. J Mol Evol, 1995, 40(3): 249-259. DOI: 10.1007/BF00163230. [5] Hué S, Clewley JP, Cane PA, et al. HIV-1 pol gene variation is sufficient for reconstruction of transmissions in the era of antiretroviral therapy[J]. AIDS, 2004, 18(5): 719-728. DOI: 10.1097/00002030-200403260-00002. [6] 伦玉华, 罗皓, 方晓君, 等. 东莞市HIV-1流行株pol基因序列特征和亚型分析[J]. 中华疾病控制杂志, 2018, 22(12): 1297-1299. DOI: 10.16462/j.cnki.zhjbkz.2018.12.022.Lun YH, Luo H, Fang XJ, et al. Subtype and sequence analysis of pol gene among HIV-1 strains in Dongguan[J]. Chin J Dis Control Prev, 2018, 22(12): 1297-1299. DOI: 10.16462/j.cnki.zhjbkz.2018.12.022. [7] Santos AF, Soares MA. HIV Genetic Diversity and Drug Resistance[J]. Viruses, 2010, 2(2): 503-531. DOI: 10.3390/v2020503. [8] Njouom R, Pasquier C, Sandres-Sauné K, et al. Assessment of HIV-1 subtyping for Cameroon strains using phylogenetic analysis of pol gene sequences[J]. J Virol Methods, 2003, 110(1): 1-8. DOI: 10.1016/s0166-0934(03)00080-6. [9] Kessler HH, Deuretzbacher D, Stelzl E, et al. Determination of human immunodeficiency virus type1 subtypes by a rapid method useful for the routine diagnostic laboratory[J]. Clin Diagn Lab Immunol, 2001, 8(5): 1018-1020. DOI: 10.1128/CDLI.8.5.1018-1020.2001. [10] Yahi N, Fantini J, Tourres C, et al. Use of drug resistance sequence data for the systematic detection of non-B human immunodeficiency virus type 1 (HIV-1) subtypes: how to create a sentinel site for monitoring the geneticdiversity of HIV-1 at a country scale[J]. J Infect Dis, 2001, 183(9): 1311-1317. DOI: 10.1086/319859. [11] Xu HT, Colby-Germinario SP, Asahchop EL, et al. Effect of mutations at position E138 in HIV-1 reverse transcriptase and their interactions with the M184I mutation on defining patterns of resistance to nonnucleoside reverse transcriptase inhibitors rilpivirine and etravirine[J]. Antimicrob Agents Chemother, 2013, 57(7): 3100-3109. DOI: 10.1128/AAC.00348-13. [12] 霍青青, 李庆海, 刘思宇, 等. 中国部分地区HIV-1 CRF01_AE流行情况[J]. 中国病毒病杂志, 2019, 9(3): 236-241. DOI: 10.16505/j.2095-0136.2019.0017.Huo QQ, Li QH, Liu SY, et al. Prevalence of HIV-1 CRF01_AE strain in partial area of China[J]. Chin J Viral Dis, 2019, 9(3): 236-241. DOI: 10.16505/j.2095-0136.2019.0017. [13] 钟敏, 杨壁珲, 杨绍敏. 云南地区123例艾滋病抗病毒治疗失败病人的HIV-1基因耐药突变位点分析[J]. 中华检验医学杂志, 2013, 36(1): 53-58. DOI: 10.3760/cma.j.issn.1009-9158.2013.01.014.Zhong M, Yang BH, Yang SM. Analysis of genotype resistance mutations sites among 123 antiretroviral-therapy failure in HIV/AIDS patients in Yunnan Province[J]. Chin J Lab Med, 2013, 36(1): 53-58. DOI: 10.3760/cma.j.issn.1009-9158.2013.01.014. [14] Li X, Li W, Zhong P, et al. Nationwide trends in molecular epidemiology of HIV-1 in China[J]. AIDS Res Hum Retroviruses, 2016, 32(9): 851-859. DOI: 10.1089/AID.2016.0029. [15] 杨绍敏, 樊移山, 李惠琴, 等. 云南省德宏和昆明地区高效抗逆转录病毒治疗后HIV-1流行毒株pol区基因变异分析[J]. 中华检验医学杂志, 2011, 34(4): 315-320. DOI: 10.3760/cma.j.issn.1009-9158.2011.04.006.Yang SM, Fan YS, Li HQ, et al. Investigation of pol gene variation of HIV-1 epidemic strains after treatment with HARRT at Dehong prefecture and Kunming in Yunnan Province[J]. Chin J Lab Med, 2011, 34(4): 315-320. DOI: 10.3760/cma.j.issn.1009-9158.2011.04.006. [16] Chen X, Zhou YH, Ye M, et al. Burmese injecting drug users in Yunnan play a pivotal role in the cross-border transmission of HIV-1 in the China-Myanmar border region[J]. Virulence. 2018, 9(1): 1195-1204. DOI: 10.1080/21505594.2018.1496777. [17] Feng Y, Takebe Y, Wei H, et al. Geographic origin and evolutionary history of China's two predominant HIV-1 circulating recombinant forms, CRF07_BC and CRF08_BC[J]. Sci Rep, 2016, 6(1): 19279. DOI: 10.1038/srep19279. [18] Zai JJ, Liu HZ, Lu ZZ, et al. Tracing the transmissiondynamics of HIV-1 CRF55_01B[J]. Sci Rep. 2020, 10(1): 5098. DOI: 10.1038/s41598-020-61870-x. [19] 柳兴凤, 李志坚, 方星, 等. 2013—2017年贵州省艾滋病患者HIV亚型分布[J]. 中华疾病控制杂志, 2019, 23(12): 1523-1526. DOI: 10.16462/j.cnki.zhjbkz.2019.12.017.Liu XF, Li ZJ, Fang X, et al. Distribution of HIV subtypes in HIV/AIDS patients in Guizhou from 2013 to 2017[J]. Chin J Dis Control Prev, 2019, 23(12): 1523-1526. DOI: 10.16462/j.cnki.zhjbkz.2019.12.017. [20] Zheng X, Tian C, Choi KH, et al. Injecting drug use and HIV infection in southwest China[J]. AIDS, 1994, 8(8): 1141-1147. DOI: 10.1097/00002030-199408000-00017. [21] Zhang L, Wang YJ, Wang BX, et al. Prevalence of HIV-1 subtypes among men who have sex with men in China: a systematic review[J]. Int J STD AIDS, 2015, 26(5): 291-305. DOI: 10.1177/0956462414543841. [22] 李敬云. 艾滋病病毒1型CRF01_AE毒株的过去、现在和将来[J]. 中华流行病学杂志, 2016, 37(4): 443-449. DOI: 10.3760/cma.j.issn.0254-6450.2016.04.001.Li JY. Vision on the past, present and future of HIV-1 CRF01_AE[J]. Chinese Journal of Epidemiology, 2016, 37(4): 443-449. DOI: 10.3760/cma.j.issn.0254-6450.2016.04.001. [23] Kandathil AJ, Ramalingam S, Kannangai R, et al. Molecular epidemiology of HIV[J]. Indian J Med Res, 2005, 121(4): 333-344. [24] 邢辉, 梁浩, 万卓越, 等. 中国CRF01-AE亚型人类免疫缺陷病毒毒株的分子流行病学研究[J]. 中华预防医学杂志, 2004, 38(5): 12-16. DOI: 10.3760/j:issn:0253-9624.2004.05.004.Xing H, Liang H, Wan ZY, et al. Distribution of recombinant human immunodeficiency virus type-1 CRF01_AE strains in China and its sequence variations in the env V3-C3 region[J]. Chin J Prev Med, 2004, 38(5): 12-16. DOI: 10.3760/j:issn:0253-9624.2004.05.004. [25] 杨洪, 叶黎, 苏玲, 等. 2011—2015年四川省男男性行为人群HIV-1新发感染率及流行趋势分析[J]. 中华预防医学杂志, 2019, 53(3): 327-329. DOI: 10.3760/cma.j.issn.0253-9624.2019.03.018.Yang H, Ye L, Su L, et al. Ananalysis on incidence of HIV-1epidemics among men who have sex with men in Sichuan Province during 2011-2015 [J]. Chin J Prev Med, 2019, 53(3): 327-329. DOI: 10.3760/cma.j.issn.0253-9624.2019.03.018. 期刊类型引用(3)
1. 何松美,梁金虎,钟庆杨,曹焕焕,尹菊侦,王福祥. 东莞市HIV-1流行株pol基因序列特征分析. 新发传染病电子杂志. 2023(06): 9-13 .  百度学术
百度学术2. 马春花,司冰倩,吴玉灵,李宗倍,裴建新,吴忠兰. HIV感染者或AIDS患者抗病毒治疗后耐药位点动态变化情况. 宁夏医科大学学报. 2023(12): 1253-1258 . 百度学术3. 段星,张志一,王继宝,叶润华,杨锦,周素娟,王译葵,杨涛,杨跃诚,唐仁海,何纳,丁盈盈,段松. 云南德宏傣族景颇族自治州2017—2019年新报告HIV感染者基因亚型分布特征分析. 上海预防医学. 2022(09): 835-841 . 百度学术其他类型引用(2)
-
 下载:
下载:
 点击查看大图
点击查看大图
计量
- 文章访问数: 1056
- HTML全文浏览量: 447
- PDF下载量: 90
- 被引次数: 5